
When you pick up a prescription at the pharmacy and see a cheaper version of a brand-name drug, you’re holding a product that went through one of the most carefully regulated processes in modern medicine. The FDA doesn’t just approve generics because they’re cheaper - it approves them because they work the same way. The legal foundation for this is the Hatch-Waxman Act of 1984, a law that changed how America gets its medicine. It created a shortcut - not a backdoor - for generic drugs to enter the market without repeating every single test done by the original drugmaker. That shortcut is called the Abbreviated New Drug Application (ANDA).
How the Hatch-Waxman Act Made Generics Possible
Before 1984, generic drug companies had to run full clinical trials just like the original brand. That meant spending hundreds of millions of dollars and waiting years to bring a copy to market. Most couldn’t afford it. So patients paid high prices, and competition was nearly nonexistent. The Hatch-Waxman Act changed that. It said: if a generic drug has the same active ingredient, strength, dosage form, and route of administration as the brand, and it delivers the same amount of medicine into your bloodstream at the same speed, then you don’t need to retest safety and effectiveness. You just need to prove bioequivalence.
This law didn’t lower standards - it made them smarter. The FDA still requires every generic to meet the same quality, purity, and strength standards as the brand. The difference? The generic company doesn’t have to prove the drug works from scratch. It can rely on the FDA’s own prior findings. That’s why today, 9 out of 10 prescriptions filled in the U.S. are for generics. That’s not luck. It’s design.
What the FDA Actually Checks Before Approval
When a generic manufacturer submits an ANDA, the FDA doesn’t just rubber-stamp it. They dig deep. Here’s what they look for:
- Active ingredient: Must be identical in chemical structure to the brand drug. No exceptions.
- Strength and dosage form: If the brand is a 20mg tablet taken orally, the generic must be the same. No smaller pills, no liquid versions unless approved.
- Bioequivalence: This is the core. The generic must deliver the same amount of active ingredient into your bloodstream within the same time frame as the brand. Typically, this is tested in 24 to 36 healthy volunteers using blood samples taken over hours. The FDA requires the generic’s absorption rate to be within 80% to 125% of the brand’s. That’s not a guess - it’s science.
- Inactive ingredients: These can be different. Fillers, dyes, preservatives - they don’t affect how the drug works, so they’re allowed to vary. But they still must be safe and meet FDA purity standards.
- Manufacturing: The factory where the generic is made must pass the same inspection as the brand’s. No corners cut. No lax quality control. The FDA shows up unannounced.
- Labeling: The generic’s label must match the brand’s in all clinically important details - uses, warnings, dosing, storage.
These aren’t suggestions. They’re requirements. And the FDA enforces them with precision. In 2023 alone, the agency approved 90 first-time generic drugs after full review. That’s not a trickle - it’s a steady flow.
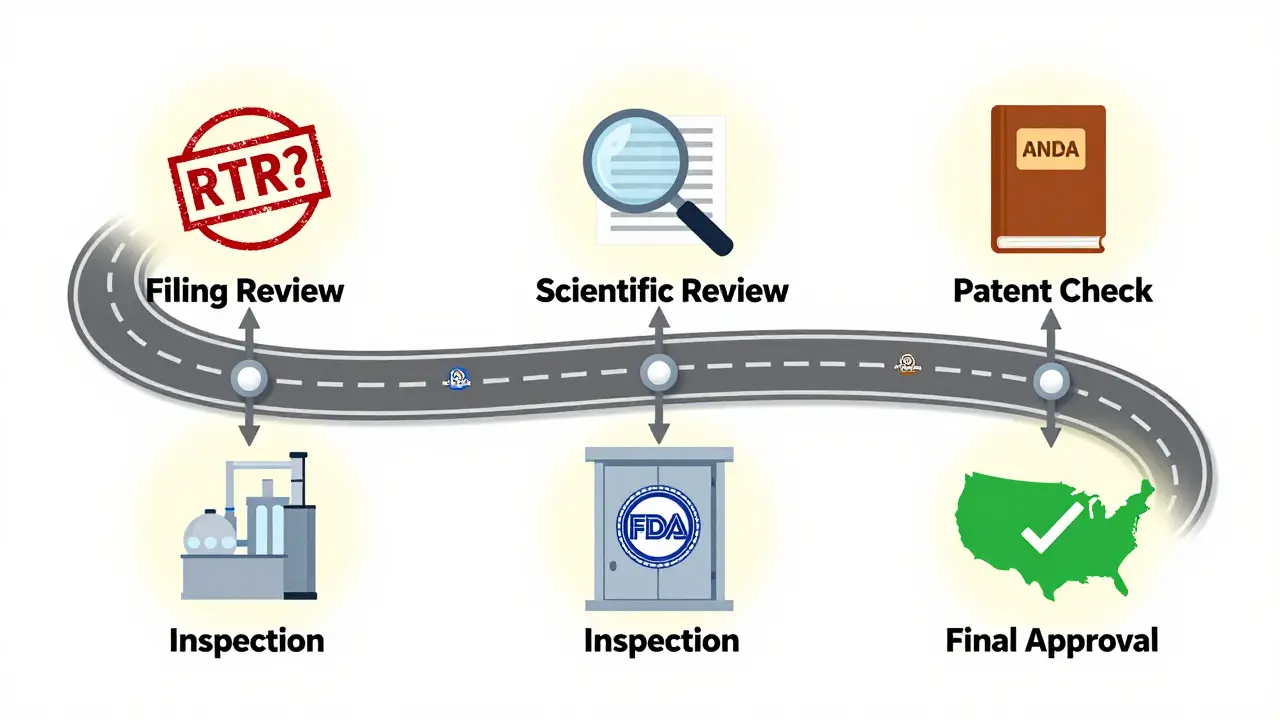
The ANDA Review Process: Step by Step
Submitting an ANDA isn’t like filling out a form. It’s a complex, multi-stage process:
- Filing Review: The FDA checks if the application is complete. If it’s missing key data - like bioequivalence studies or manufacturing details - they issue a Refuse-to-Receive (RTR) letter. The applicant must fix it, pay a new fee, and resubmit.
- Scientific Review: Once filed, the application gets assigned to a review team. They evaluate chemistry, manufacturing, bioequivalence, and labeling. This phase is governed by the Generic Drug User Fee Amendments (GDUFA), which sets strict timelines.
- Patent and Exclusivity Check: The FDA reviews the Orange Book - a public list of approved drugs, their patents, and exclusivity periods. If the brand still holds patent protection, the generic applicant must certify whether they’re challenging it. If they do (a Paragraph IV certification), the brand company can sue, triggering a 30-month legal stay. During that time, the FDA can’t approve the generic.
- Inspection: The FDA inspects the manufacturing facility. If it fails, approval is delayed until the issue is fixed.
- Final Decision: If everything checks out, the FDA approves the ANDA. The generic can then be sold in the U.S.
Under GDUFA, the FDA targets a 10-month review for standard applications and 8 months for priority ones - like drugs in shortage or first generics. That’s a huge improvement from the 180-day timeline originally written into Hatch-Waxman. The system is faster, but not rushed.

Why Some Generics Take Longer - and Why Complex Drugs Are Harder
Not all drugs are created equal. A simple tablet like lisinopril or metformin is easy to copy. But what about an inhaler? A topical cream? A long-acting injection? These are called complex generics. They’re harder to replicate because their effectiveness depends not just on the drug, but on how it’s delivered - the size of particles, the texture of the cream, the pressure of the spray.
For these, bioequivalence studies aren’t enough. The FDA needs additional data: how the drug behaves in the lung, how it penetrates the skin, how it’s released over time. The agency has launched initiatives like the Complex Generic Drug Product Development Resources to help manufacturers navigate this. But progress is slow. Many of these products still face long delays - sometimes years - before approval.
That’s why patients waiting for a generic version of a life-saving inhaler or acne cream can get frustrated. The science is harder. The testing is more expensive. And the FDA has to be extra cautious.
Cost, Competition, and Impact
Generic drugs save patients and the healthcare system billions. The average generic costs 80% to 85% less than the brand-name version. In 2022, the U.S. generic drug market was worth about $125 billion. That’s not just corporate profit - it’s access. A diabetic who can’t afford insulin may switch to a generic. A veteran on blood pressure meds might finally afford their daily dose. The FDA estimates that generics prevent over 150 million doctor visits and hospitalizations each year by making treatment affordable.
But competition matters. If only one generic manufacturer enters the market, prices don’t drop as much. The FDA’s Drug Competition Action Plan targets products with limited competition, pushing for faster approvals and clearer patent rules. The goal? More than one generic. More than one manufacturer. More than one price option.
What’s New in 2025? Faster Reviews for U.S.-Made Generics
In October 2025, the FDA announced a new pilot program: faster review times for generic drugs that are both tested and manufactured in the United States. This isn’t just about speed - it’s about supply chain security. After years of relying on overseas factories, the U.S. government is pushing to bring more drug production home. If a company builds its facility in Ohio or Georgia and tests its product here, it gets priority review. It’s a small incentive, but it could change how generics are made.
This move signals a bigger shift: the FDA isn’t just approving generics anymore. It’s trying to reshape how they’re made.
Who Makes These Generics?
Major players like Teva, Sandoz, and Viatris dominate the market. But there are hundreds of smaller companies too - some specializing in hard-to-make injectables, others in niche pediatric formulations. The FDA doesn’t favor big names. It evaluates each application on its own merits. A small company with a solid ANDA has just as good a shot as a giant.
Why This System Works - and Where It Still Struggles
The ANDA system works because it’s balanced. It protects patients by requiring strict equivalence. It protects innovation by respecting patents. And it protects affordability by cutting unnecessary costs. But it’s not perfect. Patent thickets - where brand companies file dozens of minor patents to delay generics - still slow things down. Complex drugs remain a bottleneck. And not every manufacturer has the expertise to navigate the paperwork.
Still, the numbers speak for themselves. Over 90% of prescriptions filled are generics. That’s not because people are settling. It’s because the system works. The FDA doesn’t cut corners. It just cuts the fluff.
Are generic drugs really as effective as brand-name drugs?
Yes. The FDA requires generics to have the same active ingredient, strength, dosage form, and route of administration as the brand. They must also prove bioequivalence - meaning they deliver the same amount of medicine into your bloodstream at the same rate. Studies show generics perform just as well as brands in real-world use. The FDA approves over 90% of generic drugs on their first review cycle.
Why do some generics look different from the brand?
The active ingredient is the same, but inactive ingredients - like dyes, fillers, or coatings - can differ. These affect color, shape, or taste, but not how the drug works. The FDA allows these differences as long as they don’t impact safety or effectiveness. If you’re concerned, ask your pharmacist - they can confirm the generic meets all FDA standards.
Can a generic drug be approved before the brand’s patent expires?
Only if the generic manufacturer challenges the patent with a Paragraph IV certification. If the brand company sues, the FDA must wait 30 months before approving the generic - even if the application is otherwise complete. This is called a 30-month stay. It’s a legal tool, not a regulatory one. Some generics get approved before the patent ends if the patent is found invalid or not infringed.
How long does it take for the FDA to approve a generic drug?
Under the Generic Drug User Fee Amendments (GDUFA), the FDA targets 10 months for standard applications and 8 months for priority ones - like first generics or drugs in shortage. Some applications take longer due to incomplete submissions, manufacturing issues, or patent disputes. But the system is faster and more predictable than it was before GDUFA was implemented in 2012.
Are generic drugs made in the same facilities as brand-name drugs?
Yes, and no. The FDA requires all manufacturing sites - whether for brand or generic drugs - to meet the same quality standards. Many generic manufacturers use the same facilities as brands, especially for complex drugs. Others use separate plants, but they’re still inspected to the same level. The FDA doesn’t distinguish between brand and generic facilities during inspections. Quality is non-negotiable.
The next time you fill a prescription and see a lower price tag, remember: behind that generic drug is a 40-year-old law, a rigorous science-based process, and thousands of inspections - all designed to make sure what you’re taking is safe, effective, and affordable. The system isn’t flawless, but it’s one of the most successful public health policies in modern history.







The Hatch-Waxman Act was a masterstroke in regulatory engineering-creating a scientifically rigorous yet economically viable pathway for bioequivalence validation. The ANDA framework doesn’t cut corners; it eliminates redundant preclinical and clinical trials by leveraging the FDA’s existing safety database. Bioequivalence thresholds (80–125% AUC and Cmax) aren’t arbitrary-they’re statistically anchored to clinical non-inferiority. This is pharmacokinetics as public policy.
The FDA approves generics with the same rigor as brand drugs-period. No compromises. No shortcuts. No exceptions. This isn’t a loophole-it’s a meticulously designed system that saves lives and billions annually. The fact that 90% of prescriptions are generics isn’t luck-it’s proof the system works. And anyone who says otherwise hasn’t read the Orange Book.
Interesting how the bioequivalence standards are based on real-world pharmacokinetic data. I wonder if there’s any data on whether patients actually notice differences between brand and generic in long-term use? Not asking because I doubt it-just curious if any studies tracked adherence or outcomes over time.
I’ve worked in community pharmacies for over a decade, and I’ve seen patients switch from brand to generic every day. The fear? Always there. The outcome? Almost always the same. I’ve had diabetics tell me they couldn’t afford insulin until generics hit. That’s not just policy-it’s dignity. The FDA’s system isn’t perfect, but it’s one of the few things that actually works for people who need it most.
If you’re still skeptical about generics, ask yourself this: why do hospitals, the VA, and Medicare all use them as first-line? Because they’re safe. Because they’re effective. Because they’re cheaper without sacrificing quality. This isn’t about saving money-it’s about saving access. Every time you choose a generic, you’re voting for a healthcare system that works for everyone-not just the wealthy.
India produces 60% of the world’s generic drugs and we do it better than anyone else. The FDA is finally waking up to the fact that quality doesn’t come from America alone. Our manufacturers meet USP standards, pass FDA inspections, and deliver life-saving meds at 1/5th the cost. Why are we still importing from China? We could make all of this here. We have the talent. We have the infrastructure. We have the will. It’s time the FDA stopped treating Indian manufacturers like second-class citizens.
One must ask: if the FDA permits bioequivalence as a surrogate for clinical efficacy, does this not represent a dangerous epistemological shift? We are replacing empirical, patient-level outcomes with pharmacokinetic proxies. This is not science-it is technocratic reductionism. The patient is no longer the unit of analysis; the plasma concentration curve is. And in this system, the human being becomes a variable in a statistical model.
Have you ever wondered if the FDA is being manipulated by Big Pharma? Think about it-patent extensions, Paragraph IV challenges, 30-month stays… it’s all a game. The system looks fair, but the rules are written by lawyers, not scientists. And those ‘unannounced inspections’? I’ve heard stories-facilities get warned weeks in advance. How do I know? My cousin works in QC for a ‘generic’ company. She says they prep like it’s a military inspection. And the ‘inactive ingredients’? Sometimes they cause reactions. No one talks about that.
Let’s be real-the ANDA system is a beautiful machine, but it’s got grease under the hood. Companies game the Orange Book like it’s a board game. Patent thickets? More like patent jungles. And don’t get me started on the ‘complex generics’ loophole-where a $200 inhaler becomes a $1,200 ‘new drug’ because the propellant is 0.3% different. The FDA’s ‘Complex Generic Drug Product Development Resources’? Cute name. Still takes three years to approve. Meanwhile, patients are dying waiting for a generic asthma pump that should’ve been out in 2021.
My mom takes a generic blood pressure med. She’s been on it for 8 years. No issues. No side effects. She says it’s the same as the brand. I asked her if she could tell the difference. She said, ‘I just take the pill. I don’t care what it’s called.’
It’s amazing how many people still think generics are ‘second-rate.’ The truth? They’re often made in the same factories, under the same inspections, by the same engineers. The only difference? The label. And the price tag. That’s not a compromise-it’s a victory for public health. We should be celebrating this system, not doubting it.
Generics saved my dad’s life. He couldn’t afford the brand. Now he’s on a $3 generic. He’s still here. That’s all that matters. 🙏
The real innovation isn’t just the ANDA-it’s the GDUFA timeline. Before 2012, reviews took 18–24 months. Now? Under 10. That’s not minor-it’s transformative. And the new pilot for U.S.-made generics? That’s the next frontier. Bringing production home isn’t just about security-it’s about accountability. When the factory is in Ohio, not Shanghai, the FDA can actually show up. And that’s how you build trust.